三代测序在病毒基因组测序中已有较出色的表现,但准确率方面还需进一步的提升。尽管现有的Polish工具取得了令人满意的结果,但仍有改进纠错性能的空间,以实现更精确的基因组组装。
本文介绍一款在病毒RNA编码区的Polish软件,可对已知RNA病毒组装前和组装后的序列进行Polish。软件文章HMMPolish:a coding region polishing tool for TGS-sequenced RNA viruses发表在Briefings in Bioinformatics (IF=9.5)上,可下载阅读。
该软件需要安装以下依赖软件:
Biopython:conda install conda-forge::biopython
HMMER:conda install bioconda::hmmer
pandas:conda install conda-forge::pandas
上述软件安装完成后,安装HMMPolish,这里直接下载即可:
git clone--recursive https://github.com/rainyrubyzhou/HMMPolish HMMPolish
cd HMMPolish/src
python HMMPolish.py -h
下载过程中可能遇到“Empty reply from server”等字样,不用担心,多试几次就好了!
python HMMPolish.py –h后出现帮助文档信息,表明软件完成安装,可以正常使用。
需要在python3.0以上,经过尝试在python2.7中无法使用。
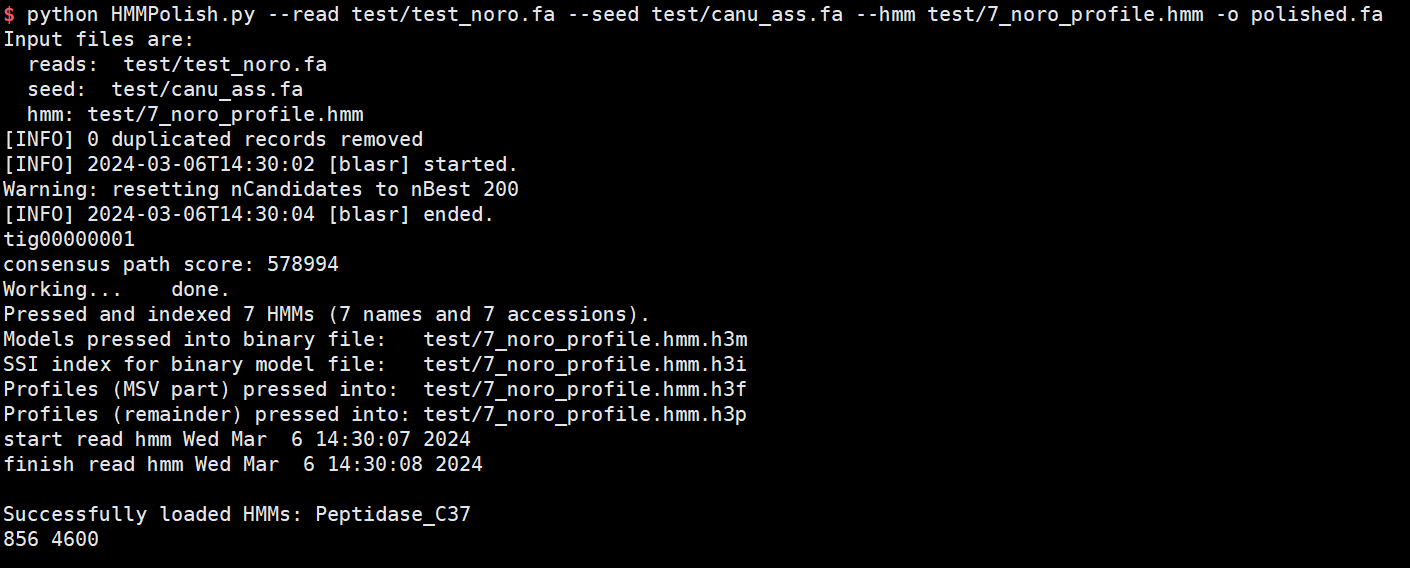
python HMMPolish.py--read test/test_noro.fa--seed test/canu_ass.fa –hmm test/7_noro_profile.hmm -o polished.fa
polished.fa的文件即为编码区域polish之后的文件。
--read:输入的reads文件;
--seed:可以是组装软件组装出来的序列,也可以是原始的序列;
--hmm:.hmm格式的配置文件。
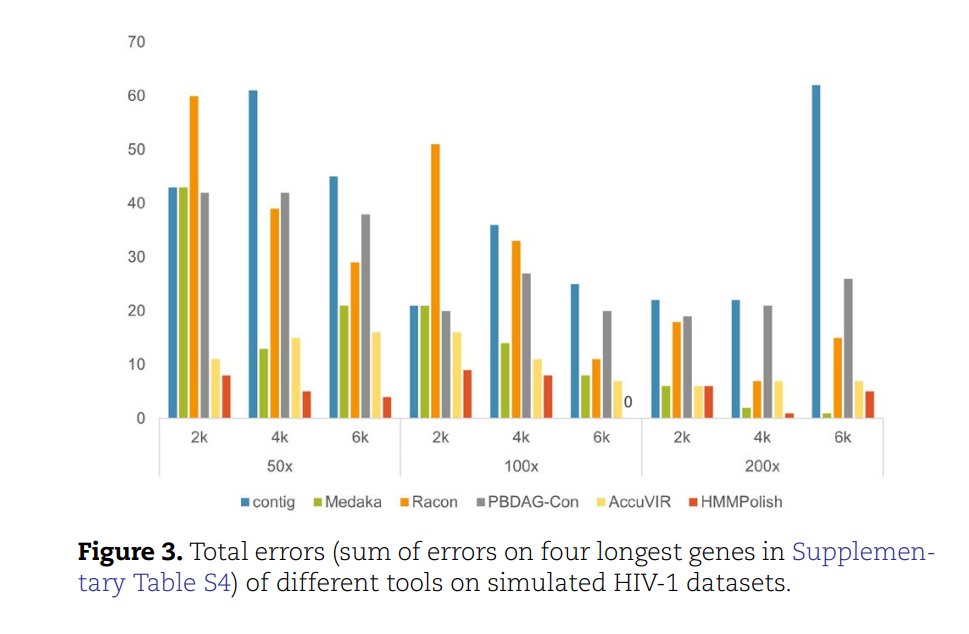
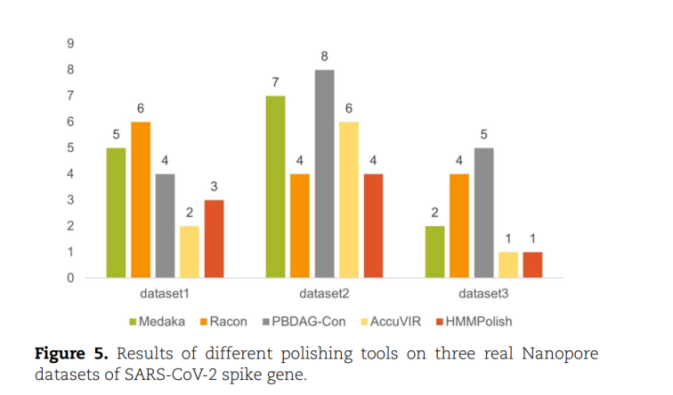
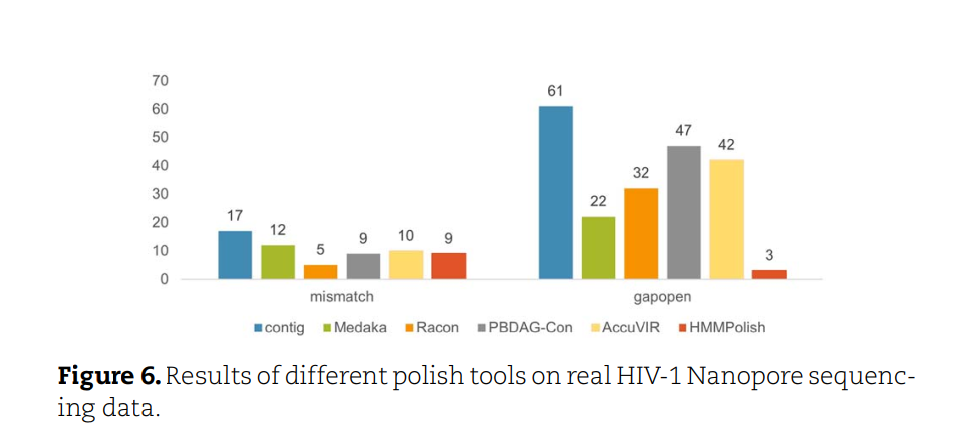
与多个Polish软件进行对比时,还是展现出较好的纠错能力[1],如果大家感兴趣,可以在自己的科学研究中进行尝试。此外,该软件也有一定的局限性,目前只能应用在部分已知的病毒中。
[1] Yu R, Abdullah SMU, Sun Y. HMMPolish: a coding region polishing tool for TGS-sequenced RNA viruses. Brief Bioinform. 2023 Sep 20;24(5):bbad264.