随着测序技术的不断发展,尤其是近些年崛起的长读长测序,产生了大量的长读长序列,如何高效率地将这些reads比对到参考序列上,成了人们关注的重要问题。
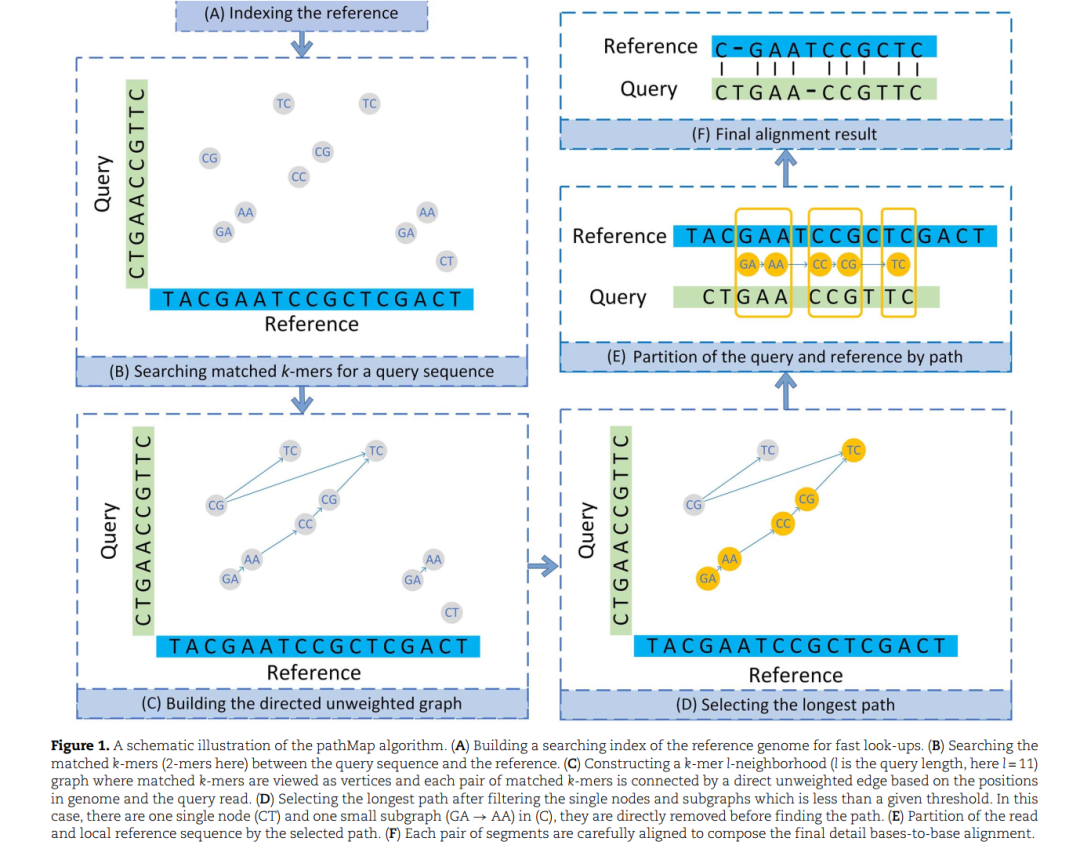
本文介绍一款专门应用于单分子测序single-molecule sequencing(SMS)产生的序列比对分析工具,具有鲁棒性(robust)强,灵敏度(sensitive)高等优势。软件文章pathMap: a path-based mapping tool for long noisy reads with high sensitivity发表在Briefings in Bioinformatics(IF=9.5)上,可下载阅读。pathMap独有特征是通过最长的path来选择最佳比对的链。
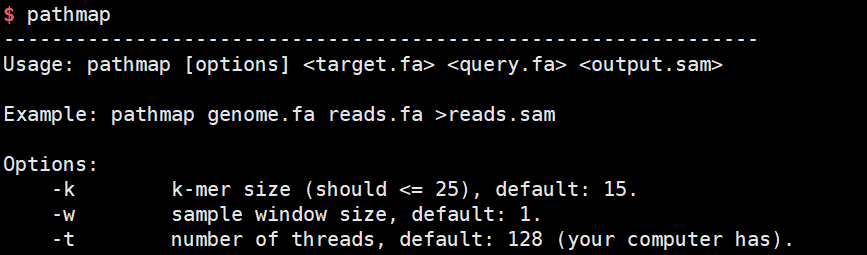
运行命令:使用默认参数进行演示
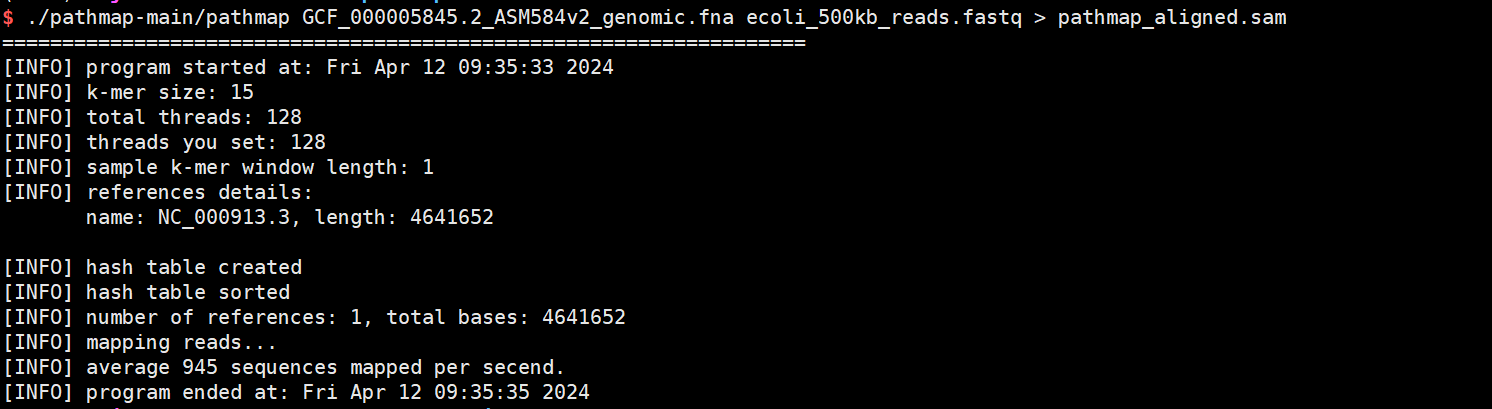
./pathmap-main/pathmap GCF_000005845.2_ASM584v2_genomic.fna ecoli_500kb_reads.fastq > pathmap_aligned.sam
pathmap_aligned.sam即比对的sam文件,可用于后续的分析。
与其他类型的比对工具进行比较:
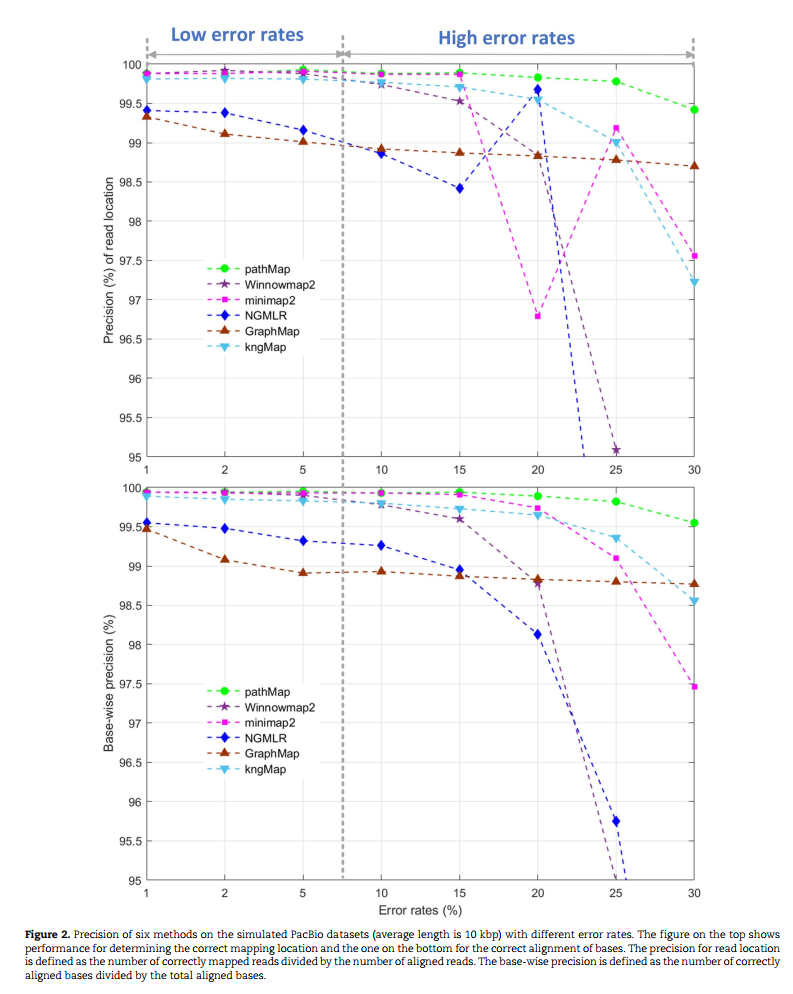
测序的纠错可靠性更强:无论在低错误率区还是在高错误率区,序列的mapping都有稳定而准确的表现;
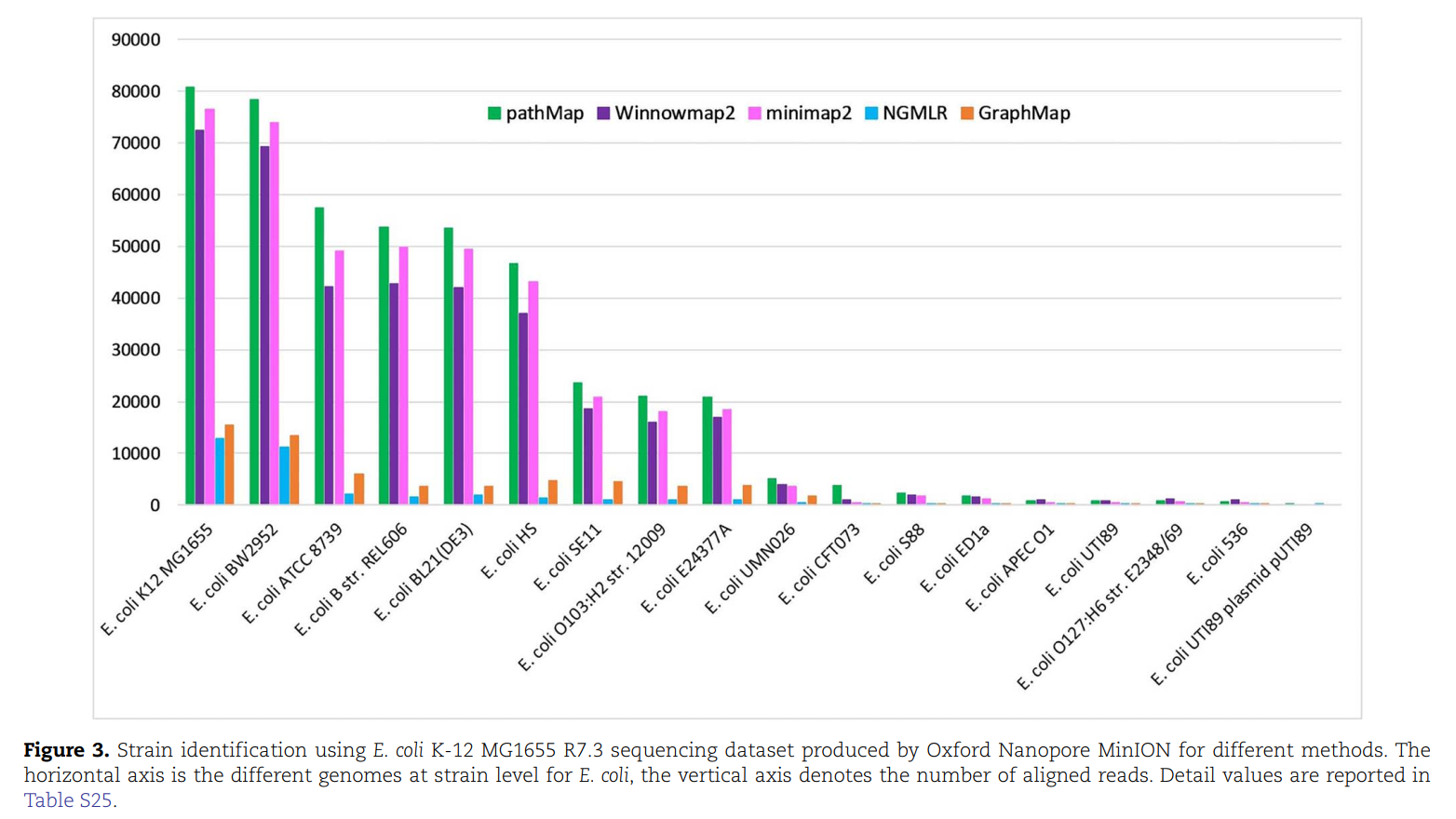
在对病原微生物的种属鉴定种中有更高得灵敏度;
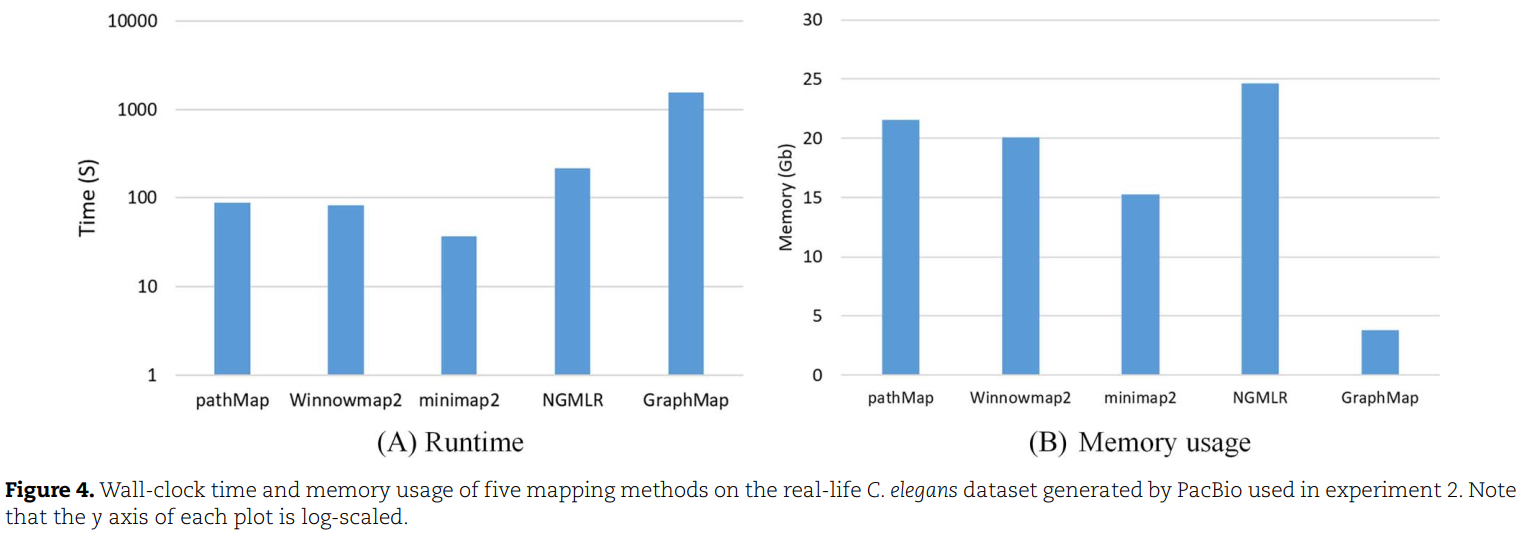
运行速度和内存使用:pathMap运行速度较快,占用内存与Winnowmap2相当。
[1] Wei ZG, Zhang XD, Fan XG, Qian Y, Liu F, Wu FX. pathMap: a path-based mapping tool for long noisy reads with high sensitivity. Brief Bioinform. 2024 Jan 22;25(2):bbae107.
[2] https://github.com/zhang134/pathmap.git