短串联重复序列(Short tandem repeats,STRs)是人类基因组中一类丰富的结构或功能元件,在人类群体中表现出重复序列的长度和遗传变异的多态性。在短读长测序无法准确识别STRs的问题上,以长读长测序的新型检测方法展现出了较强的优势,在处理计算STRs重复次数上更加需要合适的软件工具进行分析,本文将以Nanopore三代测序数据进行软件使用介绍。
1、简介
NanoRepeat是一款准确定量分析简单类型(single tandem repeat)和两种连接类型(adjacent repeats)的短串联重复序列工具,具有使用简单,运行速度快等特点。
2、软件安装
该软件需要在Python(version >= 3.8)的环境里运行,这是许多生物软件使用时,值得注意的地方。另外,还需要安装好Minimap2(version >= 2.22)的比对软件和处理bam和sam文件的Samtools (version >= 1.13)软件。
Minimap2:conda install -c bioconda minimap2
Samtools:conda install -c bioconda samtool
上述软件安装完成后,继续搭建:
*生物信息学分析软件要重点关注环境的搭建,很多都是依赖不同版本的python,否则会出现报错运行不了的情况。
conda create -n nanorepeat python=3.8 #“Proceed ([y]/n)” 选择“y”即可
conda activate nanorepeat #根据安装末尾提示,激活软件运行环境
git clone https://github.com/WGLab/NanoRepeat.git #下载NanoRepeat目录
cd NanoRepeat
pip install . #安装,出现Successfully installed字样,安装成功
3、软件使用
下载示例数据,数据解压缩:
wget https://github.com/WGLab/NanoRepeat/releases/download/v1.3/NanoRepeat_v1.3_example_data.tar.bz2
tar xjf NanoRepeat_v1.3_example_data.tar.bz2
运行命令:
nanoRepeat.py –i HG002/hg002_Q20.20210805_3flowcells.hs37d5.example_regions.bam –t bam –d ont_q20 –r HG002/GRCh37_chr1.fasta –b HG002/ HG002_GRCh37_example_regions.bed –c 8 –o ./nanorepeat_output/HG002


出现Program finished字样,表明结束运行。
参数详解:
-i:指定需要分析数据的目录位置 ;
-t:可以作为分析的数据有4种:bam,cram,fastq和fasta;
-d:数据类型分为5种:
其中基于ONT纳米孔测序的有:1. ont_q20:Q20试剂;2. ont_sup:R9芯片配合超高精准模式;3. ont:R9芯片配合快速模式和高精准模式
基于PacBio测序有:1. hifi:HiFi/CCS模式;2. clr:连续长读长模式
默认使用ont模式
-r:参考序列
-b:记录串联重复序列的位置信息标准文件,包含4列信息:chromosome;start_position;end_position;repeat_unit_sequence
*需要注意的是bed文件中起始位置和终止位置
-c:使用CPU数
-o:输出目录
结果展示:
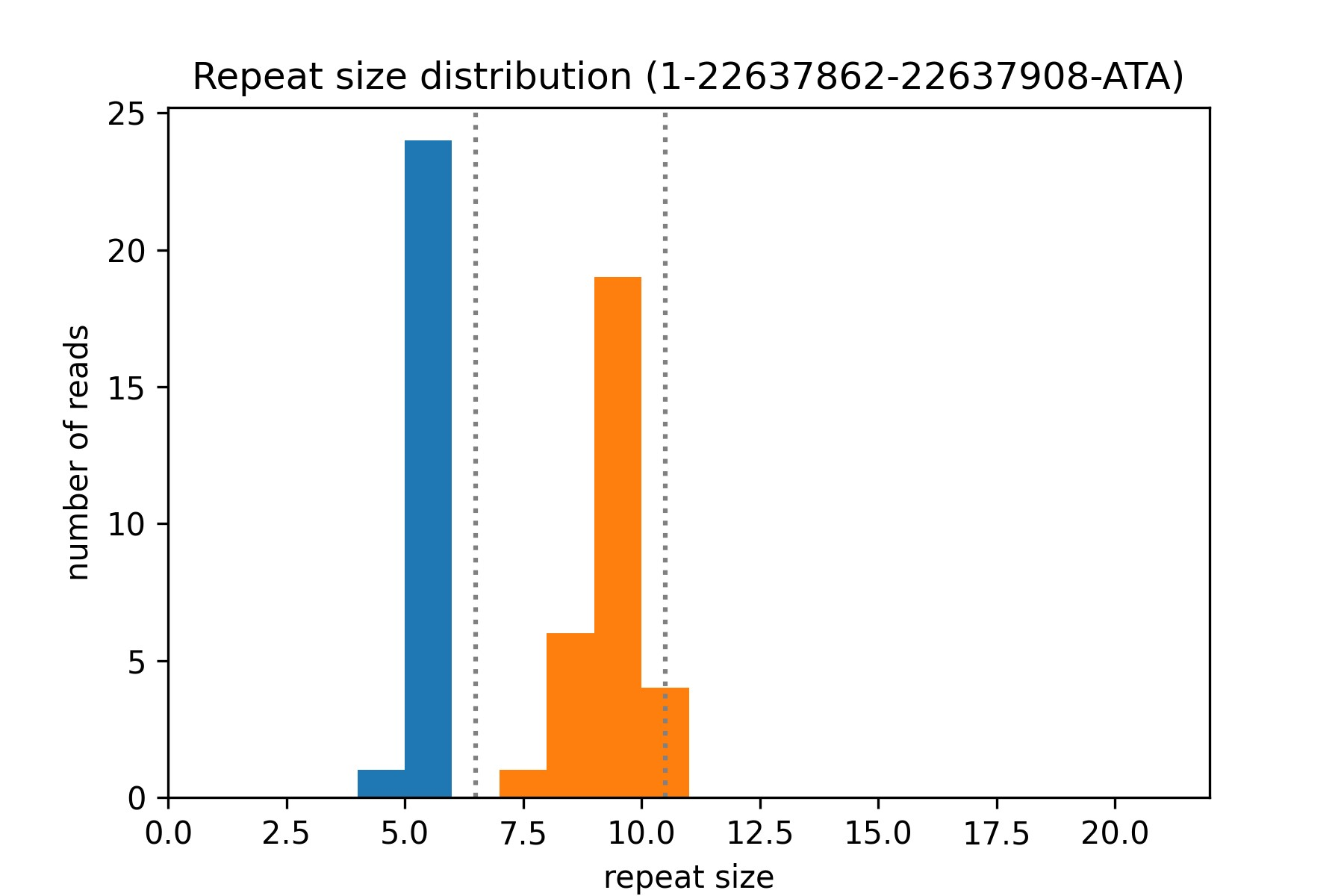
结果图中的两种颜色表示不同的allele;横坐标表示不同的重复次数,纵坐标为每种类型repeat对应的测序reads数。
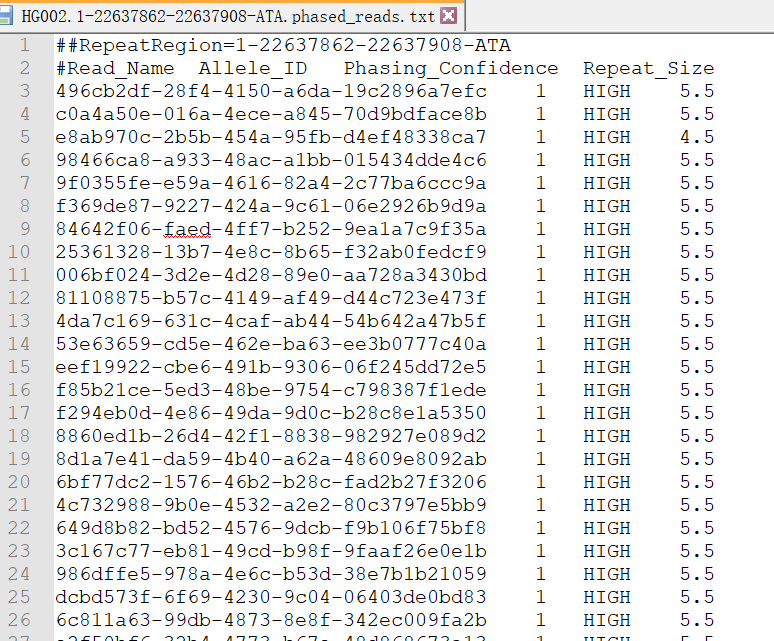
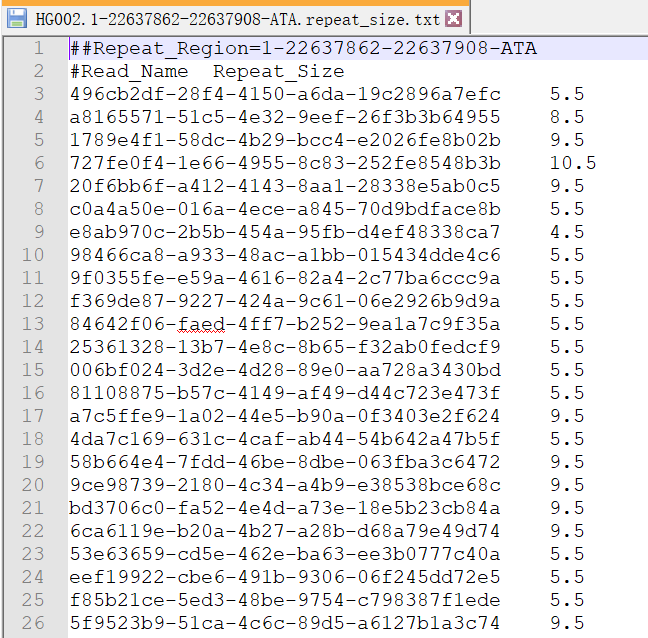
具体的重复序列描述统计保存在. repeat_size.txt和. phased_reads.txt文件中。
4、特殊类型STRs量化识别
如adjacent STRs,人类基因HTT的重复类型为(CAG)m-CAA-CAG-CCG-CCA-(CCG)n
nanoRepeat-joint.py –i HTT_amplicon/HTT_amplicon.fastq.gz –r HTT_amplicon/GRCh38_chr4.0_4Mb.fasta -1 chr4:3074876:3074933:CAG:200 -2 chr4:3074946:3074966:CCG:20 –o ./joint_quantification_output/HTT –c 8

所用到的参数和上述3类似,区别在于使用nanoRepeat-joint.py模块进行识别;位置参数-1和-2来指定重复序列的位置。
在结果中同样是用柱形图展示出每种重复类型的次数,以及mapping到的reads数。
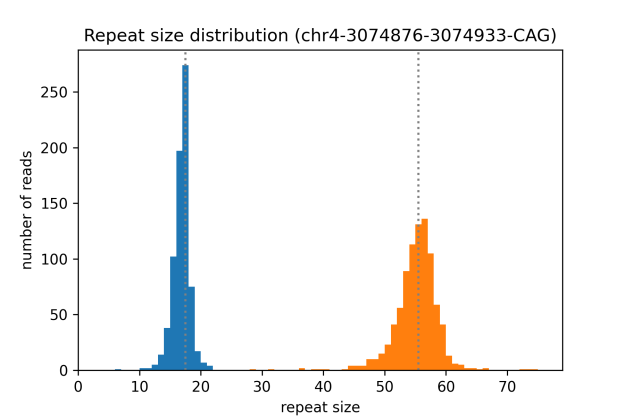

另外,可以将两种重复类型在同一个热图中展示,结果更直观。
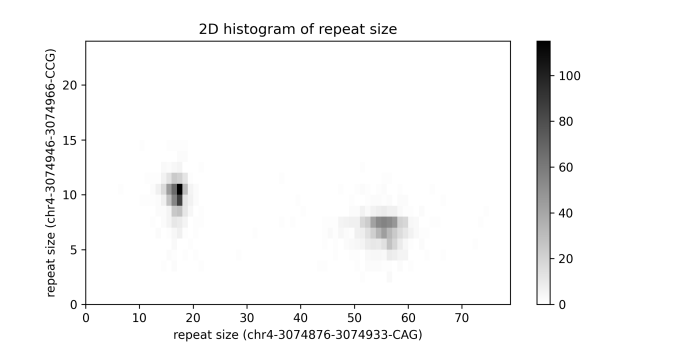
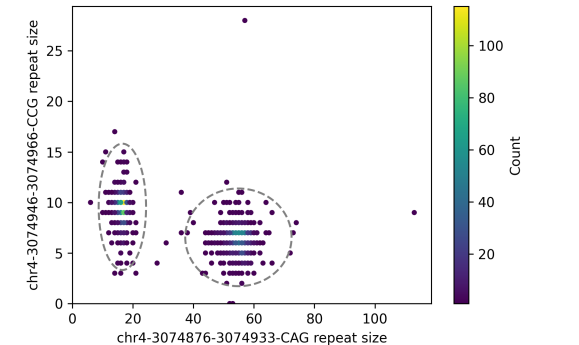
结果文件.phased_reads.txt和. repeat_size.txt中同样是统计各类型重复的详细情况。
[1] Fang L, Monteys AM, Dürr A, Keiser M, Cheng C, Harapanahalli A, et al. Haplotyping SNPs for allele-specific gene editing of the expanded huntingtin allele using long-read sequencing. Human Genetics and Genomics Advances. 2023;4(1):100146.
DOI: https://doi.org/10.1016/j.xhgg.2022.100146.
[2] https://github.com/WGLab/NanoRepeat
[3] Fang L, Monteys AM, Dürr A, Keiser M, Cheng C, Harapanahalli A, Gonzalez-Alegre P, Davidson BL, Wang K. Haplotyping SNPs for allele-specific gene editing of the expanded huntingtin allele using long-read sequencing. HGG Adv. 2022 Sep 26;4(1):100146.



