在对测序数据进行分析时,经常会碰到fasta文件的处理,如基因组组装、RNA-seq和蛋白质组学的分析等。使用现有的分析工具如SeqKit统计长度,往往需要手动筛选序列,再利用R或Python进行分析数据的可视化。这无疑是会导致操作的冗余且容易出错。本文介绍一款处理fasta数据的分析软件——SeqLengthPlot,其可将序列长度分析全流程(统计→筛选→可视化)整合为单一命令行工具,显著提升效率。
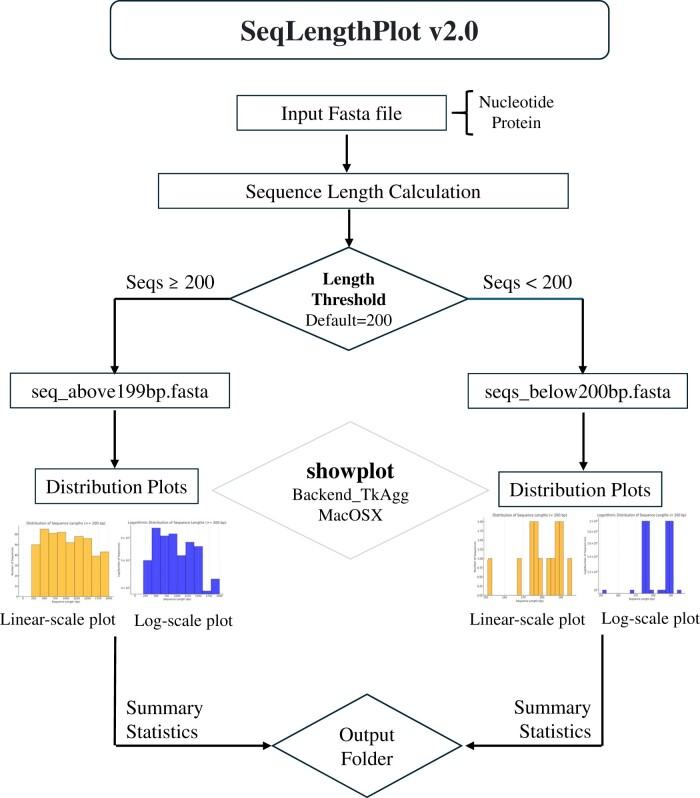
SeqLengthPlot是一种一体化序列长度分析与可视化工具,其主要功能是可对fasta文件进行统计。此外,还可对序列进行自定义长度的拆分,得到两种长度的序列文件(“长序列”和“短序列”)。同时,在分析过程中可进行直方图,直观展示数据特征。
git clone https://github.com/danydguezperez/SeqLengthPlot.git
python SeqLengthPlot_v2.0.2.py -i input.fasta
python SeqLengthPlot_v2.0.2.py -i Assembly_Ss_SE.Trinity.fasta -o output --cutoff 1000 –nt --showplot
发现直接执行上述命令会发生错误
原因是TkAgg是Matplotlib 的交互式后端,依赖 tkinter(Python的GUI 库)。在 Linux 服务器等无显示器的环境中,GUI不可用,导致此错误。这里解决的方案是使用非交互式后端(如Agg),并执行以下命令:
python SeqLengthPlot_v2.0.2.py -i Assembly_Ss_SE.Trinity.fasta -o output --cutoff 1000 --nt --showplot --backend Agg
成功,无报错信息,获取结果文件。
基本选项:
✔ -h, --help:显示帮助信息;
✔ -i INPUT, --input INPUT:必选参数,指定输入FASTA文件的路径;
✔ -o OUTPUT, --output OUTPUT:指定输出文件的目录。如果没有提供,默认使用输入文件所在的目录。
序列处理选项:
✔ --cutoff CUTOFF:设置分割序列的长度阈值(默认值为200);
✔ --nt:指定输入文件包含核苷酸序列(默认选项);
✔ --prot:指定输入文件包含蛋白质序列。
可视化选项:
✔ --showplot:启用交互式显示图表(默认不显示);
✔ --backend BACKEND:设置绘图的后端引擎(默认是 TkAgg),对于Mac用户,可以指定 MacOSX 以获得更好的兼容性。
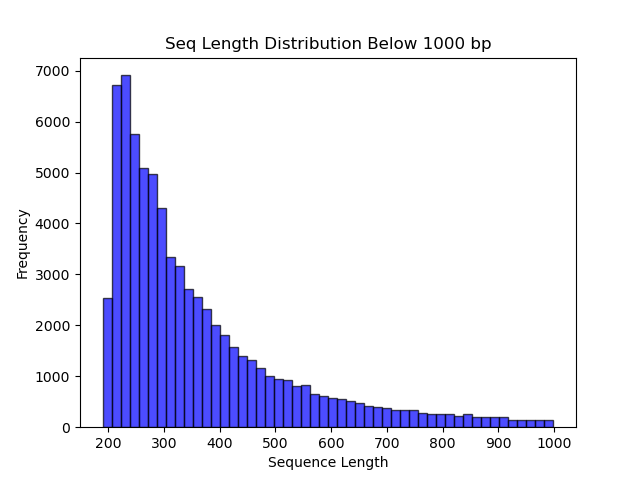
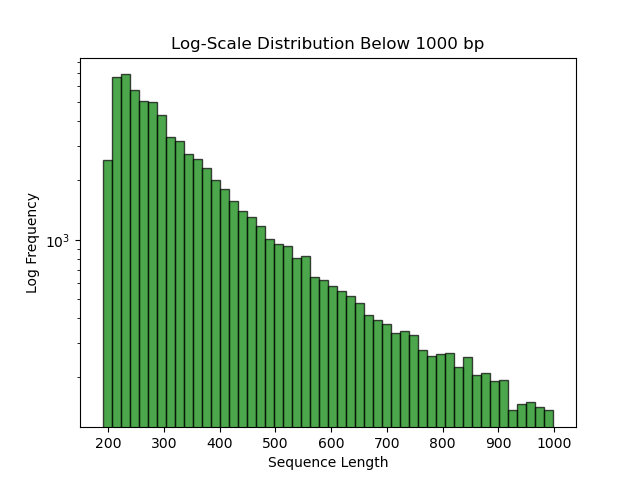
以上为自定义长度≥1000bp和<1000bp序列长度分布图(PNG 格式)。
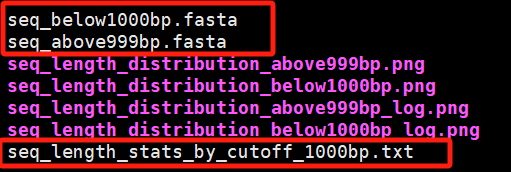
在结果文件中,还包含长度 ≥1000bp和<1000bp拆分后的fasta文件,seq_length_stats_by_cutoff_1000bp.txt文件可快速获取数据集的统计摘要。
参考文献
[1] Domínguez-Pérez D, Agüero-Chapin G, Leone S, Modica MV. SeqLengthPlot v2.0: an all-in-one, easy-to-use tool for visualizing and retrieving sequence lengths from FASTA files. Bioinform Adv. 2024 Nov 20;5(1):vbae183.
[2] https://github.com/danydguezperez/SeqLengthPlot